Đã khá lâu từ bài viết cuối cùng của mình trên Viblo, nguyên nhân là do chuyên tâm vào đồ án tốt nghiệp. Mở màn cho sự trở lại bùng nổ, mình xin phép chia sẻ về việc phân tích một số định dạng file trong Tin sinh học – một môn tương đối mới mẻ về việc áp dụng trí tuệ nhân tạo vào sinh học. Nội dung này cũng là một phần trong đồ án tốt nghiệp của mình, rất mong nhận được sự đóng góp ý kiến cũng như ủng hộ từ mọi người!
Một vài khái niệm liên quan
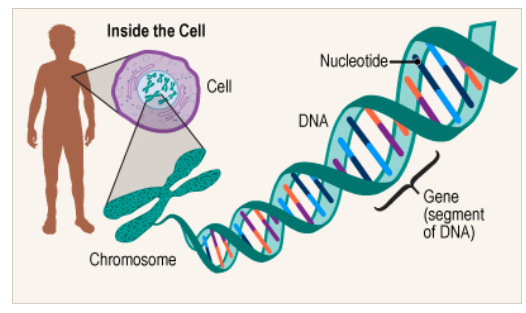
1. Gene
- Con người được hình thành từ hàng nghìn tỷ tế bào (Cell)
- Nhiễm sắc thể (Chromosome) là vật thể di truyền tồn tại trong nhân tế bào. Con người có 23 cặp NST trong đó 22 cặp NST giống nhau ở nam và nữ, cặp thứ 23 quyết định giới tính, gọi là NST giới tính
- DNA là vật liệu di truyền của tế bào, lưu trữ trong các nhiễm sắc thể ở nhân tế bào và ty thể. DNA gồm 2 chuỗi xoắn kép với nhau. Chuỗi xoắn kép đó thực chất là 1 phân tử rất dài, chỉ gồm 4 nguyên tử cơ bản cấu tạo nên : A (Adenine), T (Thymine), G (Guanine) và C (Cytosine)
- Gen, đơn vị cơ bản của di truyền, là một đoạn DNA chứa tất cả các thông tin cần thiết để tổng hợp một polypeptide (protein). Con người có khoảng 20.000 đến 23.000 gen tùy thuộc vào cách xác định gen.
- Bộ gen tham chiếu được xuất phát từ dự án Human Genome Project được khởi động vào năm 1990 bởi Viện Y tế Quốc gia và Bộ năng lượng của Mỹ nhằm xác định trình tự các cặp cơ sở trên toàn bộ gen con người. Do hạn chế về công nghệ của thế kỉ 20, các nhà khoa học đành chọn giải pháp chọn ngẫu nhiên một người tương đối khoẻ mạnh về mặt lâm sàng làm chuẩn, các nghiên cứu sau này sẽ dựa trên sự khác biệt của từng người với bộ chuẩn đó. Tất cả các khác biệt của loài người so với bộ gen này, sẽ đều được goi là “biến thể”. Bộ gen tham chiếu liên tục được cập nhật theo hướng ngày càng chính xác hơn, tổng hợp được nhiều thông tin hơn để biểu diễn khái quát nhất cho bộ gen của loài người.
2. Các loại đột biến gen
- Đột biến thay thế: Đột biến làm thay đổi một cặp bazơ DNA dẫn tới việc tạo ra một axit amin mới thay thế axit amin ban đầu được tạo ra bởi gen
- Đột biến vô nghĩa: Đột biến cũng làm thay đổi một cặp cơ sở DNA nhưng thay vì tạo ra axit amin thay thế cho axit amin ban đầu thì đoạn trình tự DNA thay thế sẽ báo hiệu tế bào ngừng xây dựng protein. Loại đột biến này làm cho protein bị ngắn đi, có thể khiến hoạt động không đúng hoặc hoàn toàn không hoạt động
- Đột biến chèn: Đột biến làm thay đổi số lượng các cặp cơ sở trong gen bằng cách thêm một đoạn DNA, kết quả là protein do gen tạo ra có thể hoạt động không bình thường
- Đột biến xóa: Đột biến này cũng làm thay đổi số lượng các cặp cơ sở DNA nhưng bằng cách loại bỏ đi một đoạn DNA. Sự xóa bỏ nhỏ có thể xóa bỏ một hoặc một vài cặp cơ sở trong một gen, trong khi đột biến mất đoạn lớn có thể xóa bỏ toàn bộ một gen hoặc một vài gen lân cận. DNA bị xóa có thể làm thay đổi chức năng của protein được tạo thành từ gen đó
- Đột biến nhân bản: một đoạn DNA bị sao chép một cách bất thường một hoặc nhiều lần. Dạng đột biến này có thể thay đổi chức năng của protein tạo thành
- Đột biến lệch khung: Đột biến xảy ra khi DNA bị chèn thêm hoặc mất đi các cặp cơ sở làm thay đổi khung đọc của gen (khung đọc chứa bộ 3 mã hóa, mỗi bộ 3 mã hóa cho một axit amin). Đột biến khung làm thay đổi nhóm các bazơ và thay đổi mã hóa cho các axit amin. Protrin tạo thành thường không có chức năng. Đột biến chèn, xóa và đột biến nhân bản đều là những đột biến dịch khung.
- Đột biến lặp lại mở rộng: Một đoạn DNA ngắn được lặp lại liên tục nhiều lần, làm tăng số lượng nhân lên của đoạn trình tự DNA ngắn. Loại đột biến này có thể khiến protein tạo thành hoạt động không bình thường.
3. Định dạng file FASTQ
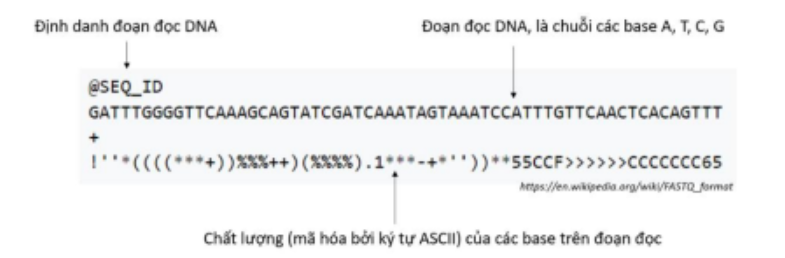
Kết quả từ máy giải trình tự gen sẽ là các đoạn đọc DNA, được cung cấp dưới dạng file FASTQ. Mỗi file FASTQ sẽ chứa số lượng đoạn đọc khác nhau tùy thuộc vào mẫu được giải trình tự, mỗi đoạn đọc đều gồm 3 phần chính:
- Định danh đoạn đọc DNA: Mỗi đoạn đọc DNA sẽ có một mã duy nhất để phân biệt
- Đoạn đọc DNA: Đoạn đọc định dạng chuỗi, bao gồm các cơ sở A, T, G, C
- Chất lượng của các cơ sở trên đoạn đọc: Gồm 1 chuỗi các kỹ tự mang ý nghĩa chất lượng của các cơ sở trên đoạn đọc, được mã hóa bởi kỹ tự ASCII. Xác suất cơ sở đó đọc bị lỗi được tính theo công thức
P=10−(Q−33)10P = 10 ^ { frac { – left ( Q – 33 right ) } { 10 } }
Trong đó, Q là giá trị theo mã ASCII của ký tự mã hóa chất lượng của cơ sở; P là xác suất cơ sở đọc được từ máy giải trình tự bị lỗi
4. Định dạng file SAM/BAM
SAM là viết tắt của định dạng Căn chỉnh trình tự / Bản đồ (Sequence Alignment/Map format). Đây là một định dạng văn bản được phân tách bằng TAB bao gồm một tiêu đề (phần này là tùy chọn) và phần căn chỉnh. Nếu có, tiêu đề phải nằm trước các căn chỉnh.Mục đích ban đầu của tệp SAM là lưu trữ thông tin ánh xạ cho các chuỗi từ trình tự thông lượng cao. Do đó, một bản ghi SAM không chỉ cần lưu trữ trình tự và chất lượng của nó mà còn cần lưu trữ thông tin về vị trí và cách thức một bản ghi trình tự đưa vào tham chiếu.
File SAM gồm 2 phần lớn:
-
Phần tiêu đề (The header section): Phần này là tùy chọn, có thể có hoặc không, phải bắt đầu với ký tự @. Phần này lưu trữ thông tin về tham chiếu mà các trình tự đã được ánh xạ tới và công cụ được sử dụng để tạo tệp SAM
![image.png]()
-
Phần căn chỉnh (The alignment section)}: Phần mang nhiều thông tin ý nghĩa nhất của File SAM, bao gồm 11 trường dữ liệu được phân tách bằng TAB. Nếu bất kì trường nào có giá trị không có sẵn, sẽ được đặt là ‘0’ hoặc ‘*’. Thông tin khái quát của 11 trường như sau:
- QNAME: ID của đoạn đọc trong file FASTQ
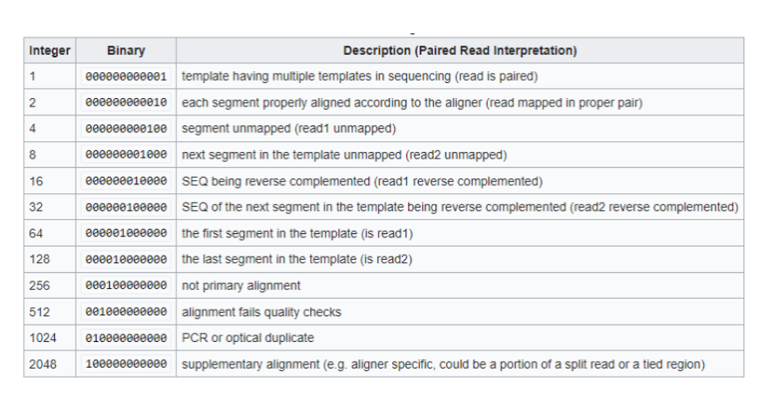
- FLAG: Trường FLAG có giá trị là 1 số nguyên dương. Để lấy thông tin của trường FLAG, cần biểu diễn dưới dạng nhị phân, ý nghĩa của bit 1 ở từng vị trí mang ý nghĩa khác nhau, được quy định như hình
![image.png]()
- RNAME: Tên của đoạn gen được tham chiếu tới (reference sequence). Trong trường hợp đoạn đọc DNA không thể ánh xạ được, RNAME mang giá trị ‘*’, ngược lại RNAME phải xuất hiện ở phần tiêu đề
- POS: Vị trí của đoạn đọc DNA trên đoạn trình tự tham chiếu (reference sequence), tính bắt đầu từ 1. Trong trường hợp đoạn trình tự đọc DNA không thể ánh xạ được, POS có giá trị bằng 0
- MAPQ: Chất lượng ánh xạ. Được tính theo công thức (Trong đó, P là xác suất của việc ánh xạ sai. ): MAPQ = -10.log(10P)
- CIGAR: viết tắt của Concise Idiosyncratic Gapped Alignment Report (CIGAR). Mang thông tin đoạn đọc DNA sau khi được so khớp với đoạn gen tham chiếu. Các ký hiệu trong CIGAR được giải thích theo hình
![image.png]()
- RNEXT: Tên của đoạn gen được tham chiếu khi căn chỉnh đoạn đọc mate-read (trong trường hợp sử dụng kỹ thuật đọc đầu cuối) hoặc đoạn đọc DNA tiếp theo sau khi đã sắp xếp theo thứ tự nếu là Single-read sequencing. Tương tự RNAME, trong trường hợp không thể ánh xạ được tới đoạn gen tham chiếu, RNEXT có giá trị ‘*’.
- PNEXT: Vị trí của đoạn mate-read hoặc đoạn đọc DNA tiếp theo sau khi đã sắp xếp theo thứ tự nếu là Single-read sequencing trên đoạn RNEXT (tính bắt đầu từ 1). Trong trường hợp đoạn đọc được không thể ánh xạ được tới đoạn gen tham chiếu, PNEXT có giá trị là 0
- TLEN: Khoảng cách giữa phần cuối được ánh xạ của mẫu và phần đầu được ánh xạ của mẫu. Mục đích của trường này là chỉ ra vị trí của đầu kia của mẫu đã được căn chỉnh mà không cần đọc phần còn lại của file SAM
- Nếu TLEN > 0: Phân đoạn ngoài cùng bên trái của mẫu
- Nếu TLEN < 0: Phân đoạn ngoài cùng bên phải của mẫu
- Nếu TLEN = 0: Mẫu 1 phân đoạn hoặc khi thông tin không có sẵn
- SEQ: Segment sequence, có chiều dài bằng hoặc nhỏ hơn so với chiều dài đã khai báo ở CIGAR. Nếu có giá trị là ‘=’ tức là đoạn đọc giống hệ với đoạn trong đoạn tham chiếu tương ứng
- QUAL: Chất lượng của đoạn đọc
- Option field: Các trường dữ liệu thông tin thêm

5. Định dạng file VCF
Variant Call Format (VCF) là định định dạng của một tệp văn bản được sử dụng trong tin sinh học để lưu trữ các biến thể trình tự gen. Thông thường, một mẫu DNA được giải trình tự thông qua hệ thống giải trình tự, tạo ra một tệp trình tự thô (Lưu trữ dạng file FASTQ). Dữ liệu chuỗi thô đó sau đó được căn chỉnh, kết quả là tạo ra các tệp BAM / SAM (BAM là dạng nhị phân của SAM). Từ đó, kết quả của việc gọi biến thể xác định những thay đổi đối với một bộ gen cụ thể so với bộ gen tham chiếu được lưu trữ trong một định dạng lưu trữ biến thể, chính là VCF. Quy trình tạo ra dữ liệu được minh họa như hình dưới
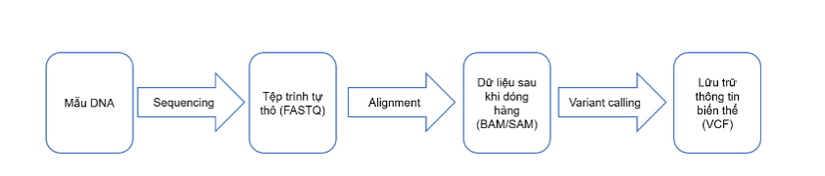
File VCF gồm 2 phần: Phần thông tin và phần dữ liệu:
-
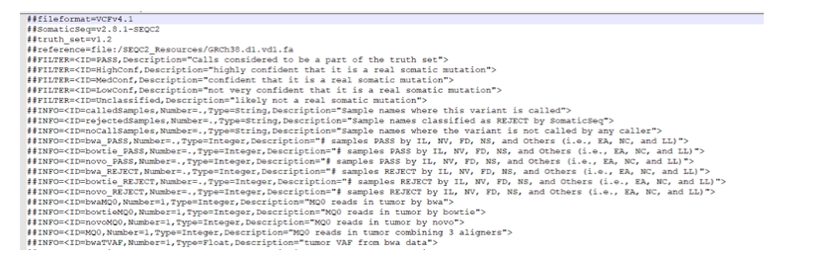
Phần thông tin: Bắt đầu bởi ##, chứa các thông tin diễn giải cho phần nội dung của file như hình
![image.png]()
-
Phần dữ liệu: Phần dữ liệu có định dạng bảng, được phân tách với nhau bởi TAB, những giá trị bị thiếu được ký hiệu bởi dấu chấm (‘.’). Dòng bắt đầu bởi ký tự # là tên của các cột. Hình dưới minh họa nội dung phần dữ liệu trong file VCF
![image.png]()

Phần dữ liệu bao gồm 8 cột thông tin bắt buộc:
- CHROM – chromosome: Tên của đoạn gen tham chiếu.
- POS – position: Vị trí trên đoạn gen tham chiếu (tính từ vị trí 1).
- ID – indertifier: Chứa ID của biến thể phát hiện được nếu có trong cơ sở dữ liệu, nếu biến thể chưa có trong cơ sở dữ liệu, giá trị ID được gán bằng dấu chấm (‘.’).
- REF – reference base(s): Alen trên đoạn gen tham chiếu.
- ALT – alternate base(s): Alen trên mẫu giải trình tự.
- QUAL – quality: Điểm chất lượng theo tỷ lệ Phred cho alen ở cột ALT.
- FILTER – filter status: Mang giá trị PASS nếu các chỉ số của biến thể thỏa mãn điều kiện do các công cụ quy định.
- INFO – additional information: Chứa các thông tin, chỉ số của biến thể phát hiện được, do các công cụ xác định biến thể tính toán ví dụ như Kiểu gen (GT – Genotype), Độ sâu giải trình tự (DP – Depth), …
Nguồn: viblo.asia